Ecco il secondo numero della raccolta di basi per affrontare in maniera “più tecnica” la spettroscopia molecolare ed in particolare parleremo delle Basi quantomeccaniche per lo studio di transizioni vibrorotazionali.
Dallo studio degli spettri vibrorotazionali di molecole è possibile trarre un elevato numero di informazioni sulla loro natura tramite la determinazione di parametri che le caratterizzano.
Fin dai livelli di istruzione generale, ci è stato detto che un generico sistema è in grado di assorbire soltanto radiazioni aventi una determinata energia (frequenza) ed in particolare solo se questa energia è uguale al salto energetico esistente fra due livelli energetici del sistema irradiato.
La teoria quantomeccanica dimostra che non tutte le transizioni sono permesse; anzi, proprio queste limitazioni (regole di selezione) permettono, almeno nel caso di molecole semplici, un’interpretazione semplice ed estremamente efficiente degli spettri.
L’interazione si può esprimere in veste matematica solo se riusciamo a risolvere l’equazione di Schrodinger:
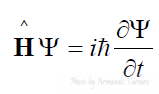
Tale equazione ammette delle soluzioni che sono la descrizione degli stati in cui un atomo, molecola può esistere.
Il simbolo H è definito operatore Hamiltoniano. Nel nostro caso l’operatore Hamiltoniano comprenderà un termine perturbativo che tiene conto dell’energia d’interazione con il campo e.m..
Il sistema, che inizialmente si trova in uno stato (stazionario) che chiameremo n, caratterizzato da una certa n, verrà perturbato per un certo tempo t fino a raggiungere un nuovo stato esprimibile come opportuna combinazione lineare dei suoi stati stazionari (teorema di
completezza):

come se l’effetto della perturbazione fosse stato quello di allontanare il sistema dal suo stato “mescolando” a questo altri stati, ognuno con un peso diverso, identificabile con |ck|2 per lo stato k –esimo.
E’ proprio a quest’ultima quantità che è proporzionale la probabilità che una data transizione possa avvenire: quanto maggiore è il valore di |ck|2, tanto più lo stato finale del sistema somiglierà al k – esimo, e quindi l’interazione e.m. avrà prodotto quella che chiamiamo una transizione dallo stato n a quello k.
Definendo il momento di transizione R nk come

il fenomeno sarà permesso o meno a secondo se tale quantità tende a 1.
Il termine è l’operatore momento di dipolo elettrico definito dalla relazione:

Essendo quest’ultimo una funzione dispari delle coordinate, affinché la quantità dkn sia non nulla, è necessario che k e n abbiano parità diversa (vedi Laporte). Per le transizioni vibrorotazionali che avvengono all’interno dello stesso stato elettronico di una molecola biatomica abbiamo (approssimazione di Born – Oppenheimer) che:

Il termine in parentesi rappresenta il momento di dipolo elettrico della molecola nello stato elettronico in esame.
Questo fatto ha un’immediata conseguenza: solo le molecole che presentano un momento di dipolo elettrico intrinseco possono esibire transizioni vibrorotazionali non accompagnate a transizioni elettroniche.
Oltre a questa condizione (necessaria), affinché una transizione vibrorotazionle sia permessa, è necessario che l’intero integrale suddetto sia diverso da
zero. Data la diversa parità delle nucl al variare di J e v (numeri quantici rotazionale e vibrazionale), tale condizione sarà verificata solo per particolari coppie di stati.
Più precisamente, si dimostra che devono essere soddisfatte le seguenti relazioni (regole di selezione specifiche):

Per comodità e precisione nell’indicazione dei simboli alleghiamo anche il testo in formato PDF.
Buona lettura:
Basi quantomeccaniche per lo studio di transizioni vibro-rotazionali
I documenti sono di proprietà del sottoscritto e non possono essere riprodotti senza autorizzazione. Prego, contattarci per maggiori dettagli.
Attached documents are my intellectual property and cannot be reproduced without my permission. Please, keep in touch with us to have more information.